HuBMAP Whole Genome Sequencing (WGS)
Last Updated 6/23/2020
Overview
This document details bulk whole genome sequencing assays, data states, metadata fields, file structure, QA/QC thresholds, and data processing.
Description
Whole genome sequencing (WGS) measures the genome-wide nucleotide sequence in a biological sample. Generally, the purpose is to screen the entire genome for all sequence variations (against a reference sequence) such as benign sequence variants (SNPs) or candidate pathogenic mutations. Examples of sequence variants include chromosomal rearrangements, nucleotide substitutions, deletions or insertions. An example use case would be a genome-wide search for somatic mutations (cancer-causing mutations that arose in a somatic cell, as opposed to a germline cell) by comparing DNA sequence in a patient’s tumor cells to that in the same patient’s healthy cells. See Appendix 1, below, for more detailed description.
HuBMAP Whole Genome Sequencing Data States (Levels)
The HuBMAP project provides data to the public in a variety of data states, which denote the amount of processing that has been done to the data. The data states for WGS seq data provided by the HuBMAP project are listed below:
| Data State | Description | Example File Type |
|---|---|---|
| 0 | Raw data: This is the raw sequence data (unprocessed) generated directly by the sequence instrument in files either with Phred quality scores (fastq). | FASTQ |
| 1 | Aligned data: SAM files contain sequence data that has been aligned to a reference genome and includes chromosome coordinates. BAM files are compressed binary versions of SAM files | SAM, BAM |
| 2 | Mutations: Variant call format (VCF). | .vcf |
HuBMAP Metadata:
-
Level 1: These are attributes that are common to all assays, for example, the type (“CODEX”) and category of assay (“imaging”), a timestamp, and the name of the person who executed the assay.
-
Level 2: These are attributes that are common to a category of HuBMAP assays, i.e. imaging, sequencing, or mass spectrometry. For example, for imaging assays this includes fields such as x resolution and y resolution.
-
Level 3: These are attributes that are specific to the type of assay, for example for CODEX that would include number of antibodies and number of cycles.
-
Level 4: This is information that might be unique to a lab or is not required for reproducibility or is otherwise not relevant for outside groups. This information is submitted in the form of a single file, a ZIP archive containing multiple files, or a directory of files. There is no formatting requirement (although formats readable with common tools such as text editors are preferable over proprietary binary formats).
All HuBMAP data will have searchable metadata fields. The metadata schema is available in Github for download.
Values to be produced by HIVE Pipeline
| Level | Field | Definition | Valid Values | Purpose |
|---|---|---|---|---|
| na | data_analysis_protocols_io_doi | Link to the protocol document describing how the HIVE or TMC is processing the data | ||
| na | reference_genome | Genome used for alignment | GRCh38 or GRCh37 | |
| na | mapping_platform | Software used for quantification | BWA-MEM | |
| na | mapping_version | Version of BWA-MEM used, with HuBMAP-specific modifications | ||
| na | number_of_raw_reads | Raw number of sequencing reads | Numeric Value | |
| na | quality_score | Average phred score of dataset | Numeric Value | |
| na | percent_unique_mapped_reads | When a set of reads are aligned with a genome, some will map in multiple locations. This indicates the percentage of reads that mapped to only one location on the genome | [0-1] | QA/QC |
HuBMAP WGS Sequence Raw File Structure
The raw sequencing data is recorded in a FASTQ file which contains sequenced reads and corresponding sequencing quality information. Every read in FASTQ format is stored in four lines as follows
@HWI-ST1276:71:C1162ACXX:1:1101:1208:2458 1:N:0:CGATGT
NAAGAACACGTTCGGTCACCTCAGCACACTTGTGAATGTCATGGGATCCAT
+
#55???BBBBB?BA@DEEFFCFFHHFFCFFHHHHHHHFAE0ECFFD/AEHH
Line 1 begins with a ‘@’ character and is followed by a sequence identifier and an optional description (such as a FASTA title line). Line 2 is the sequence of the read. Line 3 begins with a ‘+’ character and is optionally followed by the same sequence identifier (and any description) again. Line 4 encodes the quality values for the bases in Line 2.
HuBMAP QA/QC of raw (state0) data files
The bolded steps below constitute a series of standard RNA-seq data analysis workflow.
Pre-alignment QC with FastQC
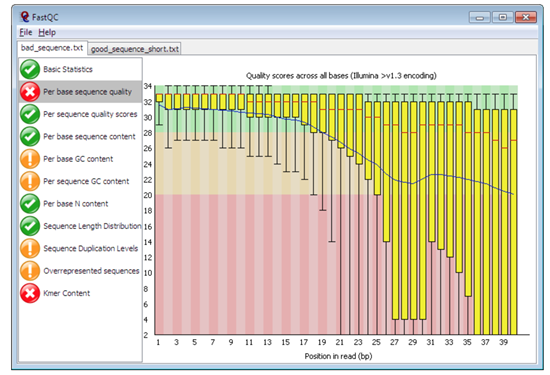 Figure 1: Plot of per sequence base quality ((Figure from Babraham Bioinoformatics)
Figure 1: Plot of per sequence base quality ((Figure from Babraham Bioinoformatics)
| qc_metric | Threshold | Method |
|---|---|---|
| average_base_quality_scores | >20 (accuracy rate 99%) | FastQC |
| gc_content | FastQC | |
| sequence_length_distribution | >45 (encode) | FastQC |
| sequence_duplication | FastQC | |
| k-mer_overrepresentation | 20 (accuracy rate 99%) | |
| contamination_of_primers_and_adapters_in_sequencing_data | Library specific data on adapters need to be provided to the read-trimming tool like trimmomatic (Bioinformatics. 2014 Aug 1; 30(15):2114-20.). |
Terms defined in this document
Base quality scores: prediction of the probability of an error in base calling GC content: Percentage of bases that are either guanine (G) or cytosine (C) K-mer overrepresentation: Overrepresented k-mer sequences in a sequencing library
Library-level Alignment QC: Note that this is not per-cell. Trimmed reads are mapped to reference genome.
| qc_metric | Threshold | Method |
|---|---|---|
| unique_mapping_percent | Ideally > 95% (Encode) Acceptable > 80% (at least for bulk) | SAMtools/Picard |
| duplicate_reads_percent | SAMtools/Picard | |
| fragment_length_distribution | >45 (encode) | SAMtools/Picard |
| gc_bias | Biased if variance of GC content is larger than 95% of confidence threshold of the baseline variance | SAMtools/Picard |
| library_complexity | NRF>0.9, PBC1>0.9, and PBC2>3 | https://www.encodeproject.org/data-standards/terms/#library |
Uniquely mapping % – Percentage of reads that map to exactly one location within the reference genome.
Duplicated reads % - Percentage of reads that map to the same genomic position and have the same unique molecular identifier (Encode)
Post-alignment processing QC: (see Per cell QC metrics table below)
-
Remove duplicated reads
-
Remove low quality reads
-
Remove mtDNA reads
Appendix 1. Brief detailed description of WGS protocol
New England Biosciences (NEB) whole genome sequencing library preparation kit is outlined below. A total amount of 1.0μg DNA per sample was used as input material for the DNA sample preparations. Sequencing libraries were generated using NEBNext® DNA Library Prep Kit following manufacturer’s recommendations and indices were added to each sample. The genomic DNA is randomly fragmented to a size of 350bp by shearing, then DNA fragments were end polished, A-tailed, and ligated with the NEBNext adapter for Illumina sequencing, and further PCR enriched by P5 and indexed P7 oligos. The PCR products were purified (AMPure XP system) and resulted libraries were analyzed for size distribution by Agilent 2100 Bioanalyzer and quantified using real-time PCR. See the detailed protocol here: dx.doi.org/10.17504/protocols.io.bfsmjnc6
This protocol adheres to the MINSEQE standards put forward by the Functional Genomics Data Society (FGED).
For Additional Help
Please contact: Aaron Horning